![[ CSPS Pharmaceuticals, Inc. ]](/csps/image/cspslogo12horizsmaller.png)
![[ SPYDER ]](/images/sponsspyder5.gif)
| | |
|
Shaking in solid phase synthesis?
Some responses to the discussion on the listserv Mol-Diversity about the necessity of shaking in the solid phase synthesis:
From Julian Hayward: Further to Matthew Todd's query about agitation/sonication, I can offer the following contribution (attempting, of course, to keep the commercial nature of this response to a minimum!).
As search in our Solid-Phase Synthesis database for the keyword 'sonication' gave 132 reactions from the following publications:
Bioorg. Med. Chem. Lett., 1995, 5, 47.
Int. J. Pept. Protein Res., 1993, 42, 138.
Int. J. Pept. Protein Res., 1994, 44, 58.
J. Am. Chem. Soc., 1976, 98, 6710.
J. Am. Chem. Soc., 1979, 101, 677.
J. Org. Chem., 1994, 59, 4723.
Synlett, 1995, 1017.
Tetrahedron Lett., 1996, 37, 3231.
Tetrahedron Lett., 1996, 37, 835.
Tetrahedron Lett., 1997, 38, 1043.
Tetrahedron, 1995, 51, 5671.
Tetrahedron, 1995, 51, 5675.
US Patent US 5324483
US Patent US 5582801
World Patent WO 9408711
The DeWitt patents, in particular, offer a wide range of reaction types.
Hope this helps.
Regards
Dr. Julian Hayward
Synopsys Scientific Systems
Voice: +44 (0)113 245 3339
Fax: +44 (0)113 243 8733
E-mail: julian@synopsys.co.uk
From Michal Lebl: Shaking can be eliminated completely if concentration of reagents allows generation of an excess inside of the polymeric matrix. In this case reaction vessels can be eliminated completely -- all reactions can be performed inside of the solid phase beads "inclusion volume chemistry". See Journal of Peptide Science, 2, 240, 1996.
Comments to Kevin Burges' letter, anyone?
I haven't seen any comments about Kevin Burgess's letter in the February 10 issue of Chemical & Engineering News. Does everyone agree with his definition of "Combinatorial Chemistry"? In a recent review in J. Chem. Inf. Comput. Sci (actually written some months ago but appears in Jan/Feb 1997 issue) I was concerned at the use of "combinatorial chemistry" as an umbrella term. I am now writing two more articles, one of them for an Encyclopedia, and I'd quite like to address this issue.
Incidentally, I hope you've all seen Stu Borman's article in the February 24 issue of Chemical & Engineering News.
Wendy Warr
Dr Wendy A Warr
Wendy Warr and Associates, 6 Berwick Court
Holmes Chapel, Cheshire CW4 7HZ, England
Tel/fax +44 (0)1477 533837
wendy@warr.com
http://www.warr.com
MOLREPS FREQUENTLY ASKED QUESTIONS
**1 Where can I obtain libraries?**
1.1 _Phage Libraries_
You can try approaching the following people and organisations:
1.1.1 pIII/6-mer, pIII/15-mer, pVIII-4/15-mer libraries:
George Smith, University of Missouri, FAX +1-573-882-0123
1.1.2 pVIII/9-mer, pVIII/9-merCys and pVIII/zinc-finger
phagemid libraries:
Alessandra Luzzago, IRBM P. Angeletti, luzzago@irbm.it
Franco Felici, IRBM P. Angeletti, felici@irbm.it
(NON-COMMERCIAL USERS ONLY)
1.1.3 Antibody scFvs with synthetic CDRs:
Greg Winter, MRC-LMB Cambridge, gw@mrc-lmb.cam.ac.uk
Contact Fiona Sait at
fs1@mrc-lmb.cam.ac.uk
(NON-COMMERCIAL USERS ONLY)
1.1.4 Commercial Sources:
Stratagene (Fab fragments in phage lambda)
Affymax
Pharmacia
Cambridge Antibody Technology
New England Biolabs (pIII/7-mer). (NEB catalog, p.166).
see also this URL ->
http://vent.neb.com/neb/phd/phd.html
pSKAN Phagemid Display System from MoBiTec via NBL (+44-1670) 733015
EZtrap Phage Display cDNA libraries-AMS Biotechnology (+44-1993) 706500
1.2 _Synthetic Peptide Libraries_
1.2.1 Commercial Sources:
Affymax
Chiron Corporation
Most of the major peptide companies will custom-synthesise a library to your requirements.
1.3 _Nucleic Acid Libraries_ (Aptamers)
1.3.1 Jack Szostak at Harvard medical school?
1.3.2 NEXUS corporation (Larry Gold)?
1.4 _Organic Chemical Libraries_
1.4.1 Affymax?
1.4.2 Parke-Davis (DIVERSOMERS)?
**2) Where can I get anti-phage antibodies?**
2.1 anti-pIII MAb from Michael Tesar
mte@gbf-braunschweig.de
2.2 anti-pIII polyclonals from GATC, a German company,
FAX +49-7531-57313 TEL +49-7531-57204
2.3 rabbit anti-M13 from Stratagene.
2.4 sheep anti M13 phage
sheep anti M13 phage biotinylated
5 Prime - 3 Prime
Boulder CO
(800)533-5703
**3) How can I make my own libraries?**
3.1 _Phage Libraries_
You can obtain suitable vectors and strains from most of the sources in 1).
For phagemid work you can buy XL-1 Blue cells from
Stratagene and M13K07 helper phage from Pharmacia.
3.2 _Synthetic Peptide Libraries_
3.2.1 Manual synthesis
Houghten's "Tea Bag" method
Geysen's "Pin" method
MULTIBLOCK method - see http://www.5z.com/csps
Any of these can be adapted to produce either support-bound or soluble libraries.
3.2.2 Automated synthesis
Chiron Corporation (Zuckermann)
Advanced Chemtech (Commercial robot for 75K sterling)
3.3 _Nucleic Acid Libraries_
3.3.1 PCR methods
3.4 _Organic Chemical Libraries_
3.4.1 Manual synthesis
3.4.2 Automated synthesis (Advanced Chemtech)
**4) How can I analyse the results of my selection?**
4.1 Insert sequencing (phage libraries)
4.2 Micropanning (Smith and Scott, Methods Enzymol. (1993) 217:228-257)
4.3 Dot-blotting (Felici et al., J. Mol. Biol. (1991) 222:301-310)
4.4 ELISA (Smith and Scott, Methods Enzymol. (1993) 217:228-257) (Dente et al. Gene (1994) 148:7-13)
4.5 Colony immunoscreening (Christian et al. J. Mol. Biol. (1992) 227, 711-718) (Felici et al. Gene (1993) 128, 21-27)
4.5 Plaque immunoscreening (Luzzago et al., Gene (1993) 128:51-57) (Felici et al., Methods Enzymol. (1996) 267:116-129)
**5) Are there any World Wide Web (WWW) sites about molreps?**
5.1 http://www.bio.net/ - The BIOSCI web site itself (MOLREPS message archive)
5.2 http://bionmr1.rug.ac.be/chemistry/overview.html - A useful chemistry site
5.3 http://www.5z.com/ (Well, you are here!) - Site for the journal MOLECULAR DIVERSITY
5.4 http://www.mrc-cpe.cam.ac.uk/imt-doc/vbase-home-page.html - Immunoglobulin v-gene database
5.5 http://molbio.info.nih.gov/molbio/desk.html - Molecular biologists desk reference
5.6 http://www.Kairos-scientific.com/ - Kairos scientific home page
5.7 http://www-lmmb.ncifcrf.gov/~toms/sequencelogo.html - Tom Schneider's Sequence Logo
5.8 http://www.ebi.ac.uk/ - The European Bioinformatics Institute (EBI)
5.9 http://www.ebi.ac.uk/primers_home.html - PCR primers database at EBI
5.10 http://aminoacid.bri.nrc.ca:1125/ - Database of building blocks for library synthesis
5.11 http://vent.neb.com/neb/phd/phd.html - PHD phage library at New England Biolabs
5.12 ftp://ftp.netcom.com/pub/qu/quincicc/maxim.html - Another library source
5.13 http://www.ebi.ac.uk/imgt/ - Database of genes of immunological importance
5.14 http://immuno.bme.nwu.edu/ - Kabat database of proteins of immunological interest
**6) Do phage absorb non-specifically to ELISA plates?**
Yes, use MaxiSorp plates for best binding.
Answer by:
Pascal Mertens
Laboratoire d'Immunologie et Microbiologie
URBM-FUNDP
61 Rue de Bruxelles
5000 Namur, BELGIUM
Q : (Peter Nestler ) During our screenings we experience quite often that we can't wash our target proteins off the selected Tentagel-beads, not even with SDS or guanidinium hydrochloride. However, we have not observed that this effect leads to false positive screening results. Did somebody experience similar problems removing proteins from beads and does have a method to dissolve the proteins from the beads? An explanation for this observation could be that the proteins precipitate between the PEG-arms of Tentagel, which would be consistent with the findings that one can "shave" the beads on the surface by digesting peptides with proteases (latest report : Proc. Natl. Acad. Sci. USA 1996, 93, 8194), although Lowe et al. report that proteins penetrate the beads completely (Mol. Diversity 1995, 1, 223) to interact with ligands. The protein precipitation within the beads would then lead to deactivation of the enzymes. (Lowe et al. only report binding but no enzymatic activity.) Did somebody look into this effect and has another explanation?
Q : Is there any software for prediction of molecular weights of library mixture?
A : Prediction of library composition (for mass spectrometrical evaluation of the mixture) can be done by using the software Peptide Companion (software for every peptide chemist)
Q : (to Sharon Dankwardt) Where did you buy 3-amino-5-hydroxybenzoic acid. It seems to be discontinued by Fluka and nobody else carries it.
A : You are correct. 3-Amino-5-hydroxybenzoic acid is no longer available from Fluka. I had a ten gram bottle when I started the project and did not know Fluka had discontinued it. After I used it up I inquired into a custom synthesis, but they were not interested so I had to make it myself by the procedure outlined in reference 4. It is not as trivial as they make it out to be in the prep.
Q : (Gerard Dijkstra
[sgerard@knoware.nl] )
My group recently started research in the field of combinatorial chemistry.
We have synthesized some peptide libraries (using split-mix method) on
Tentagel-S-NH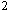 resin.
Incubation of fluorescent labeled targets (FITC or
RITC) yielded many false positive hits. We then discovered that the tentagel resin
without any peptide contains about between 0.1-2% beads with
'auto-fluorescence' properties. Extended swelling (DMF) and washing has
no effect. I wonder if more people working in this field have noticed the same.
resin.
Incubation of fluorescent labeled targets (FITC or
RITC) yielded many false positive hits. We then discovered that the tentagel resin
without any peptide contains about between 0.1-2% beads with
'auto-fluorescence' properties. Extended swelling (DMF) and washing has
no effect. I wonder if more people working in this field have noticed the same.
A : (Peter Nestler ) We have observed similar effects and did not find a way around it yet. The way we deal with the problem is to sort out the fluorescent beads before actually screening of the library. It is a tedious process and we are not 100% happy with it (also for reasons of statistics in the library composition), but it works. We are still looking for a resin that does not have these problems. We have also observed inhomogeneities in Tentagel resin: Some batches of Tentagel contained beads that interacted with our targets without any peptide attached to it, so you also want to beware of these false positives.
Q : ( M. J. Plunkett
plunkett@uclink.berkeley.edu)
Does anyone have information on suitable matrices for use in
the characterization of small molecule libraries using
MALDI mass spectrometry?
Q : ( J.
Steele)
Anyone who has worked with combinatorial libraries designed to identify
leads from screening will be aware that mixture size continues to be a
very topical issue. I wonder if subsribers to this forum ( the same
question was placed on MOLDIVERSITY listserver) have any recent insights
or opinions on the subject.
Undoubtedly, single compounds offer the best chance to get high quality data, but this must be offset against the need to test a reasonably large set of compounds (ideally a "diverse" set - whatever that means !). Many companies have settled for small mixtures containing 10 - 100 compounds to avoid high levels of apparent background activity in the search for weak leads, and this choice often seems to follow bad experience with mixture sizes in the range 500 - 1000 +.
Offset against this is the well-established view that combinatorial mixtures (as opposed to random ones used in high thoughput screening) will contain structurally related entities and so the "epitope effect" should dramatically increase the likelihood of finding a weak lead in a large order mixture. There are good examples where real peptide leads have been discovered from colossal mixtures.
Where is the balance between numbers and screening data quality ? Is the field moving toward a consensus position mixture size or are there still no guidelines ? Based on experience and literature reading, my prefernce is for mixtures of ~ 100, but I would be delighted to see some alternative preferences posted !
Comment :(M. Lebl)
Biochemists at Selectide have found that the acceptable number of compounds in the
mixture depends very much on the screened target and can be anywhere well
below 100 and well above 1000. In the situations when the mixture is
prepared for the screening against multiplicity of targets, it is better
to be conservative and work with mixture of lower number of compounds.
Pharmacopeia established their magic number to 30 compounds per well --
the reason being the shipment of libraries to several collaborating
institutions where a great variety of tests is performed.
We are preparing a small molecule library using Rink-MBHA-Polystyrene resin. We have noticed poor cleavage yields (0-30% of theoretical recovery) for many of our more electron rich products (eg. those containing indole, some phenols etc). Our current cleavage protocol is treatment with 50/45/5 TFA:DCM:water for 1 h. We suspect that these electron-rich products are cleaved from the Rink resin (carboxamide formation) but then reattach to the resin by irreversible CC bond formation (with the resin bound cation).
Has anyone else observed these low cleavage yields with electron-rich compounds?
Someone has suggested the following protocol: 5/90/5 TFA:DCM:triethyl or triisopropylsilane for 1-2 h. Would anyone care to comment on the usefulness of this or other relevant cleavage protocols?
A1 (B. Neustadt): We have observed a similar phenomenon of capture of the resin-carbonium by the ligand. We were preparing acids on SASRIN resin. This type of capture is well known to solid-phase peptide chemists. When we switched to chloro-trityl resin instead, we obtained high recovery of the same ligands from the resin. In the case of amides, a xanthenyl linker (PerSeptive or Bachem) might provide a solution to your problem.
A2 ( Michael Arlt):You should be able to check if your substrates reattach to the resin by IR. Maybe you have a clearly visible functional group in your molecule? Have you checked this? It does not help you with the problem but may be it helps you finding a solution. Do you have your compounds bound via an amide bond? Or are you trying to cleave alkylamines? This is not clear from your question and you mentioned that you are preparing a small molecule library. I am asking because we are looking for a linker for secondary amines (I have posted this as a question recently) and this resin may be a possibility.
A3 (Marcel Patek): Dr. Graybill raised an interesting question regarding insufficient compound recovery when amino acid with electron rich side chain or other electron rich moiety is cleaved from the Rink-resin. This problem was faced by peptide chemists shortly after the Rink-linker was introduced and the solution was to insert glycine between the construct and linker. We have observed the same problem when analogous compounds were cleaved from SCAL (safety-catch amide linker structurally similar to the Rink-linker) linker. We tried a variety of cleavage cocktails ranging from "mild" TFA/DCM, HF to "push-pull" methanesufonic or triflic acid/thioanisole mixtures without any reasonable improvement. The addition of triisobutylsilane (triethylsilane reduces the indole ring!) did not help much either. It scavenges efficiently pink "benzhydrylium" cation affording thus cleaner products, but it acts after the cation is formed. Furthermore, the benzhydryl skeleton is cleaved to give about 30% of dialkoxybenzylamide as a side product. The whole problem will probably be in "insufficient" protonation of carboxamide function due to the "charge dillution" caused by proximity of the electron rich aromatic system (problems were also encountered with Phe, Tyr, Trp). This seems to be not a trivial problem to solve since these linkers are based on an acidic A(Al) cleavage and the simplest recommendation would therefore be - insert the glycine. Another trick could be to "shield" the electropositive center, i.e, to replace proton by some strong Lewis acid (we did not try this one).
Anyway, good luck and let us know if you find something interesting.