Gly...).
Chapter 4.3.3 Examples of peptide formula representation acceptable to Peptide Companion
| H-Gly-Gly-Phe-Leu-OH-DCHA |
| Boc-Arg(Tos)-OPfp |
| Ac-Leu-Leu-Arg-H-[H2SO4]0.5 |
(Leupeptin hemisulfate) |
| H-Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly-NH2 |
(oxytocin - disulfide) |
| H-Cys(H)-Tyr-Ile-Gln-Asn-Cys(H)-Pro-Leu-Gly-NH2 |
(oxytocein - linear peptide) |
| Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly |
([9-1 cyclo]oxytocin - bicyclic analog) |
| H-Cys-Tyr-Ile-Glu-Asn-Cys-Pro-Lys-Gly-NH2 |
([Glu4, Lys8]oxytocin - cyclic disulfide) |
| H-Cys-Tyr-Ile-Glu(_)-Asn-Cys-Pro-Lys(_)-Gly-NH2 |
([Glu4, Lys8, 4-8 cyclo]oxytocin - bicyclic analog) |
| H-Ala-Lys(Ac-Leu-Leu)-Asp(Leu-Leu-OH)-Gly-OH |
(branched peptide) |
| [H-Gly-Pro-Phe]4-[H-Tyr-Ile-Val-Trp]4-[Lys(_)]7-Ala-OH |
(example of MAP) |
|
Chapter 5 Functions of Peptide Companion
| Figure 5 |
|
Screen Capture of Peptide
Companion
Main Window |
Software Peptide Companion is driven by standard Windows tools.
You can use either the mouse with its left button to point and click the
chosen buttons, or you can use the Tab key to move on the screen
and Alt + underlined character keys to perform the chosen task. In the
following section, all functions of the software are described.
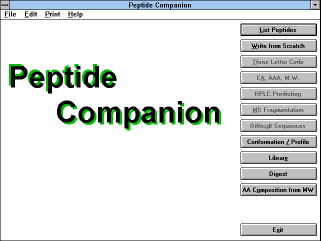
After you start the program, the
"Main" (See Figure
5) window ("Peptide
Companion") is displayed. It contains the following buttons: "List
Peptides", "Write from Scratch", "Three Letter Code", "EA, AAA,
M.W.", "HPLC Prediction", "MS Fragmentation", "Difficult
Sequences", "Conformation / Profile", "Library", "Digest", "AA
Composition from M.W." and "Exit".
Chapter 5.1 Use or modify peptide formulae (Database
of sequences, "List Peptides"
(See Figure 6) button)
| Figure 6 |
|
| Screen Capture of List Peptides
Function |
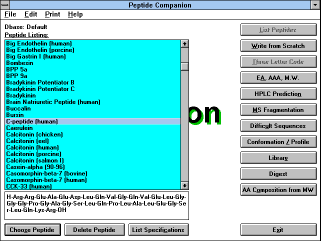
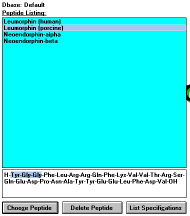
Pressing this button gives you the opportunity to choose the amino
acid sequence of the peptides stored in the database. First of all, you
can choose the database from which you will select the formula
("Default" or "Choose Dbase" buttons, if you are using a different
database than the default database, the button "Current Dbase" will
be displayed). You can also change your mind and go back and type
the formula by yourself ("Write from Scratch" button). After selection
of the database, the list of peptides in the database is displayed. You
can highlight any peptide by the mouse pointing or by using Up and
Down arrows, PgUp, PgDn, Home and End. The formula of a
selected peptide appears in the lower window. By pressing Enter or
by double clicking the mouse, or by clicking on the
"Choose Peptide" (See Figure
7)
button you load the chosen peptide for the editing.
| Figure 7 |
|
| Screen Capture of Editing
a Peptide |
If you do not intend
to edit the selected peptide, you can start the calculation using this
peptide by clicking on buttons "EA, AAA, M.W.", "HPLC Prediction",
"MS Fragmentation", "Difficult Sequences", "Conformation /
Profile". The database contains several hundred peptides, and other
peptides can be added.
Any peptide can be deleted by highlighting it
and clicking on the "Delete Peptide" button. To modify the database,
you must enter your password. The reason for this is that the peptides
in the database were very thoroughly checked and any unauthorized
modification might result in sequence information not confirmed by
CSPS or by the software licensee.
You can select all peptides from the given database fulfilling the
selection criteria.
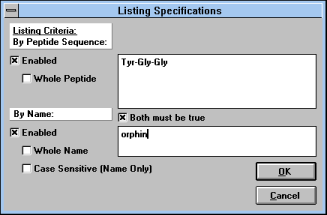
Click on "List Specifications"
(See Figure 8) and a window which
allows you to define a character string which will be sought in the
peptide formulae or in peptide names.
| Figure 8 |
|
Screen Capture of Listing
Specifications
Pop-up Window |
A search in peptide names can
be case sensitive or case insensitive, i.e. "Vaso" and "vaso" can be
different or the same -- search in peptide formulae is always case
sensitive. You can specify the search for the whole formula or name;
when this option is enabled, only fully defined items will be listed.
| Figure 9 |
|
Screen Capture of
Selected Peptide
|
You can combine both conditions in an "OR" or "AND" manner. If "Both
must be true" is checked, both conditions have to be fulfilled to give
you the positive answer. If this box is not checked, both peptides
containing the particular sequence or defined string in their names will
be listed.
Clicking on a
peptide name (See Figure
9) selected by the use of selection criteria
will display its structure in the lower field. If a structural element (partial
sequence) was used as the selection criteria, the first occurrence of
the defined sequence will be highlighted in the selected peptide
formula. If no peptide meets the selection criteria, the original
database will be displayed.
Chapter 5.2 Input formula (
"Write from Scratch" (See
Figure 10) button)
| Figure 10 |
|
Screen Capture of Write
Peptide
from Scratch |
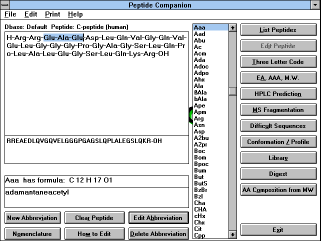
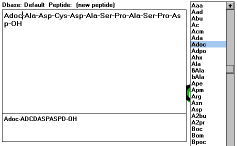
If there is no peptide sequence loaded, this button will let you type
in
the new peptide formula. An alternative way of building the formula is
double clicking on the abbreviations displayed in the window with the
list of defined abbreviations on the right. You can save the typed
peptide formula by clicking on "File" menu and selecting the "Save
Peptide" submenu. You will be given a chance to save the peptide
formula in either the database which is in use at the moment
("Current Dbase"), in any other database ("Different Dbase"), or
you can generate the new database ("New Dbase"). The computer
will then ask you for the name of the peptide. If a name you specify
already exists in the database the program will ask you to confirm
overwriting it. A formula not added to the database can be used for the
calculations, or it can be modified, but after loading another sequence
the prior one is lost. The "Edit Peptide" button is activated when a
peptide formula is loaded into the editing window. It uses the same
editing tools as defined above.
Chapter 5.3 Translation of codes ("Three Letter Code" button)
You can enter a peptide formula either in the one-letter code or in the
three-letter code. One-letter code can be entered in both the upper
(bigger) and lower (smaller) windows. Three-letter code typed in the
upper window is being translated into one-letter code in the lower
window automatically. If the formula is being typed in a one-letter code
(in any window), the button "Three Letter Code" must be clicked
before any calculation is attempted. The code is then translated into
the three letter code in the upper window. You will be warned to check
the termini of the peptide since the program is automatically assuming
that you want to calculate for an unprotected linear peptide.
Chapter 5.4 Calculation of elemental analysis, amino
acid composition and molecular weight ("EA, AAA, M.W."
(See Figure 11) button)
| Figure 11 |
|
| Screen Capture of Elemental Analysis
Window |
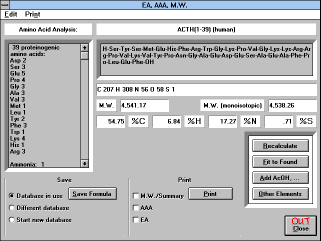
The results of the calculations are shown on the screen and can also
be sent to the printer (see section 6).
The information on the screen
shows the name of the compound, its formula, summary formula,
molecular weight, molecular weight for mass spectroscopy (M.W.
(monoisotopic)), elemental composition, and amino acid composition.
You can modify the formula in the formula window and recalculate the
result ("Recalculate" button). You can also save the peptide formula
("Save Formula" button). The "Other Elements" button will display
composition of all elements in the sample.
Chapter 5.4.1 The addition of various molecules
("Add AcOH ..." (See Figure
12) button) and calculation of probable composition of
the lyophilizate ("Fit to Found" button)
| Figure 12 |
|
| Screen Capture of Add AcOH Function
|
Workup or purification procedures of peptides result in the formation
of various salts (trifluoroacetates, hydrofluorides, hydrochlorides,
acetates, etc.). The calculation of elemental analysis of peptides with
added different molecules may show the most probable composition
of the peptide sample.
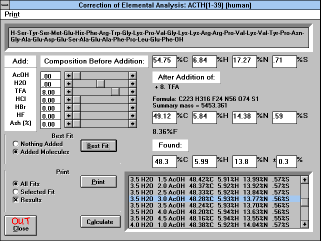
Clicking buttons "Add AcOH..." or "Fit to Found" transfers you to the
window allowing the addition of molecules of AcOH, H2O (also
subtraction), TFA, HCl, HBr, HF, and/or inorganic residues (% of Ash).
Clicking on the appropriate scroll bar can add molecules in 0.25 or
1.00 increments. Pulling the scroll bar button or entering the value in
the value window can enter any value.
Freeze-dried samples of peptides usually contain various amounts of
water and acetic acid (if this was the last solvent system from which
the peptide was lyophilized). You can try to fit the found values from
the result of elemental analysis to the theoretical values obtained by
the addition of molecules of water and acetic acid to the peptide
(either with or without added salts). You are prompted to input values
found by elemental analysis and a range in % under which you will
consider the result satisfactory. Elemental compositions satisfying the
condition are calculated. All satisfactory results are displayed in the
lower right hand corner with the best fitting analysis highlighted.
Chapter 5.5 Prediction of RP HPLC retention times
("HPLC Prediction" (See
Figure 13) button)
| Figure 13 |
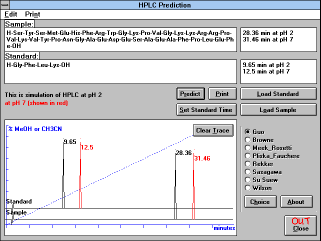
|
| Screen Capture of HPLC Prediction
Window |
You may compare the behavior of your peptide with any standard
peptide available in your laboratory. This comparison does not give
you the absolute value of k', which depends largely on the column,
instrument, and other conditions of your experiment, but it gives you
the relative elution order of two peptides at two pH values of mobile
phase. According to our experience, large deviations from the
predicted elution order usually mean that the analyzed sample has
other than expected structure (missing or redundant amino acids,
uncleaved protecting groups, modified amino acid residues). For the
best results always compare peptides of reasonably similar lengths.
Comparison of a peptide with a free amino acid is not sensible.
After pressing the "HPLC Prediction" button you will be able to
choose the prediction algorithm. You can use one of the following set
of parameters:
- Guo D., Mant C.T., Taneja A.K., Parker J.M.R. and Hodges R.S.: J.
Chromatogr. 359, 499 (1986) (This is used as the default algorithm)
- Browne C.A., Bennett H.P.J. and Solomon S.: Anal. Biochem. 124,
201 (1982)
- Meek J.L.: Proc. Natl. Acad. Sci. U.S.A. 77, 1632 (1980) and Meek
J.L. and Rossetti Z.L.: J. Chromatogr. 211, 15 (1981)
- Pliska V. and Fauchere J.L.: Proc.6th Amer.Pept.Symp., p.249.
Pierce 1979
- Rekker R.F.: The Hydrophobic Fragmental Constants, p.301.
Elsevier 1977
- Sasagawa T. and Teller D.C.: Handbook of HPLC, vol.II, p.53. CRC
Press 1984
- Su Suew J., Grego B., Niven B. and Hearn M.T.W.: J. Liq.
Chromatogr. 4, 1745 (1981)
- Wilson K.J., Van Wieringen E., Klauser S., et al: J. Chromatogr. 237,
407 (1982)
If you want to use a generic set, based on the data of Guo D., et al.
(J. Chromatogr. 359, 499 (1986)) with the correction for molecular weight
(Mant C.T., Burke T.W.L., Black J.A. and Hodges R.S.: J. Chromatogr.
458, 193 (1988)) just press the "Predict" button. This set is the most
complete since it was completed by the calculated values for various
protecting groups. If you want to use a different set of parameters,
select the desired option and press the "Predict" button again. The
new prediction will be overlayed on the trace. If you don't want to
overlay the new prediction, press "Clear Trace". The buttons "Load
Standard" and "Load Sample" will let you select the appropriate
peptide from any database of peptides available to you. Formulas are
loaded either by pressing the "OK" button or by double clicking on the
chosen name. Peptide formulas can be edited within their windows, or
they can be written from scratch. The peptide formula last edited in
the main window will be used as the sample in the transfer to the
"HPLC Prediction" window. An arbitrary peptide is used as default
standard in the prediction, unless otherwise specified.
For better comparison of calculated and experimentally determined
values, you can enter retention time of the standard peptide observed
in your experiment - - press "Set Standard Time". You will be
prompted to enter the "dead time" of your column (which depends on
the column dimensions, stationary phase, and flow rate) and retention
time of your standard (or retention times of your standard under
different conditions). Prediction will then be corrected for the values
observed experimentally. Newly entered column parameter (dead
time) will be used in all subsequent calculations during the present
session.
If you use an abbreviation, for which the retention characteristics are
not defined, you will be informed about this fact. However, the
prediction will be performed and in the case that both the standard
and your peptide contain the same unknown abbreviation, their
relative positions will be predicted correctly.
Results of predictions can be printed either as the values or also as
the traces. For the details see section 6.
Chapter 5.6 Fragmentation for Mass spectroscopy
("MS Fragmentation" (See
Figure 14) button)
| Figure 14 |
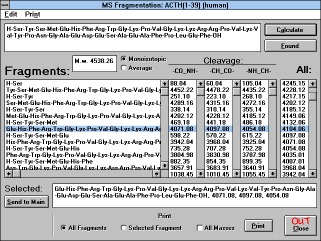
|
| Screen Capture of MS Fragmentation
Window |
This option enables you to generate probable fragments of mass
spectra of a peptide. The peptide is cleaved in all peptide bonds, and
the list of peptide fragments and a sorted list of all probable ions is
produced. However, only unbranched linear peptides give the correct
results.
Clicking on any value in the sorted list will highlight the appropriate
fragment. Double clicking on the value or fragment structure will
display its full structure and values for the appropriate possible types
of fragmentation. For more information about fragmentation of
peptides see e.g. Stults, J.T. in "Biomedical Applications of Mass
Spectrometry", Vol. 34, p. 145-201, Wiley, New York, 1990, or
Bieman, K. in "Methods in Enzymology", Vol. 193, p. 455-479,
Academic Press, New York, 1990.
| Figure 15 |
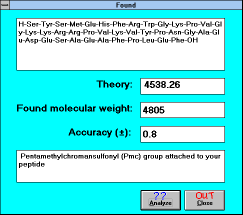
|
| Screen Capture of Analyze
Function |
The formula of the peptide can be
edited and a new fragmentation recalculated by clicking on the
"Calculate" button). The results can be presented in masses
calculated using monoisotopic values (click "Monoisotopic") for all
atoms, or using average values (click "Average").
To compare your results from the spectral measurement with the
theoretical data, click the button "Found". The displayed window will
prompt you to enter the value which you consider to be the molecular
peak of your compound. The program will analyze possible reasons
for a difference between your value and theory (by pressing the
"Analyze" (See Figure
15) button or
Enter). It will tell you whether you did not
completely remove a protecting group, modified the peptide during
work up, forgot to couple some amino acid, and so on. Remember
that this analysis depends on the precision with which you have
determined your experimental data (and which you can change by
editing the "Accuracy" window.
Chapter 5.7 Prediction of synthetically difficult
sequences ("Difficult Sequences"
(See Figure 16)button)
| Figure 16 |
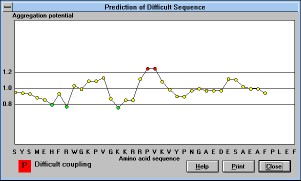
|
Screen Capture of Prediction of
Difficult
Sequences Window |
The following rules are employed for the prediction of difficult
sequences:
- (i) Coupling difficulties begin at residue number five from the
carboxy terminus. A pendent peptide chain starts to be prone to
aggregation after acquiring its fifth residue and its susceptibility to
aggregation declines with the increasing peptide length.
- (ii) The carboxy terminal six to nine residues dominantly influence
the character of the whole sequence.
- (iii) The sequence starts being potentially difficult after two or
three consecutive potentials have exceeded the value 1.1 and ends with the
same number of consecutive potentials below 0.9. With the higher
aggregation potential comes the higher proportion of pendent peptide
chains tending to aggregate. The result is the slower aminoacylation.
- (iv) In most cases aggregation profiles with seven or more consecutive
average potentials greater than 1.1 will cause difficult couplings for the
entire sequence (even if the potential drops below 0.9 for the rest of
the sequence).
- (v) Residues more than ten amino acids distant from the carboxy
terminus can be coupled without significant problems even if the
aggregation profiles show four consecutive values over 1.1 (but do not
show strong aggregation in the carboxy terminal decapeptide).
- (vi) The type of the coupled amino acid contributes significantly to
the overall aminoacylation rate. Since prediction of difficult sequences
by using the aggregation profile eliminates the effect of random coupling
problems, the relative reactivity of a coupled amino acid has to be
considered. This effect is particularly pronounced when slow reacting
amino acids are coupled to a difficult sequence.
Clicking on the "Difficult Sequences" button will show graphical
representation of aggregation potential for amino acids starting from
the fifth position in the peptide chain. If it is difficult to associate the
point on the graph with the amino acid residue on the x axis, point to
the particular position with the mouse pointer, and the amino acid
label will be displayed in the lower left corner, together with the
predicted coupling difficulty. Only sequences containing natural
unprotected amino acids can be evaluated since the aggregation
parameters are not defined for unnatural building blocks. If you want
to use this program to evaluate unnatural sequences, replace
unnatural building blocks with natural amino acids having similar
coupling properties (make a qualified guess). This program also will
not suggest the optimal combination of protecting groups to overcome
formation of difficult sequences -- there is not enough experience to
write a successful algorithm. However, once the sequence is predicted
as difficult, one should consider various synthetic alternatives for the
given sequence (various protecting groups, protected asparagine and
glutamine, protected backbone, cyclic alternatives of Cys and/or Ser,
double couplings, chaotropic salts, high temperature coupling,
coupling in ultrasonic bath, etc.).
Chapter 5.8 Evaluation of conformational parameters
or protein profile ("Conformation / Profile" button)
Chapter 5.8.1 General rules
This part of the program analyzes peptide/protein sequences. The
protein analysis includes calculation of conformational profiles
according to the Chou-Fasman algorithm and evaluation of protein
profiles and amphipathic profiles. Ten different scales are available
(hydrophilicity, hydrophobicity, acrophilicity, accessibility, antigenicity,
and hydropathy). The program uses peptide/protein sequences stored
in files in standard NIH format (see below); the sequence can be also
typed from the keyboard, edited, and stored on disk. Although this
option is intended for evaluation of longer peptides and proteins,
peptide formulas typed in the main window, or retrieved from the
database of peptides can be used. However, only peptides composed
of natural amino acids and without any side chain modifications can
be evaluated. The program will recognize any unnatural parts of the
peptide and truncate the formula.
Amino acid sequences of proteins are stored in files in standard NIH
format. Each file can contain any number of comment lines; each
comment line must start with a semi-colon (;), and all of these lines
are ignored by the program. The line immediately preceding an amino
acid sequence contains the name of the protein and it will appear on
every output as the protein name. The program uses amino acid
sequences written in the standard one-letter amino acid code using
upper- or lower-case letters. Peptide/protein sequences written in the
three-letter code will be converted to one-letter code (when entered
in the main window, or when generated by Digest option).
Standard termination of a sequence is indicated by the digit 1 or 2 or
an asterisk (1, 2, or *). Any further lines are ignored. The present
version of the program can run proteins up to 3000 amino acid
residues long.
; An example of an acceptable file. Semi-colon indicates line with comments
; First line without semi-colon contains the name of a protein
Name of protein
ACSFDGERTHNVMNSDGFARSFEGRILKSLDPLKASKNDKIEKRLNAKSLKDLKAMNVSTAFSREFDGNAMMMSNDNDKLITKLSHFGAKAMS*
Chapter 5.8.2 Functions of the
"Predict" (See Figure
17) window
| Figure 17 |
|
| Screen Capture of Predict Window |
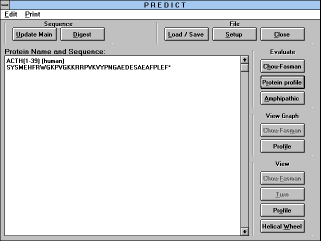
Clicking on the "Conformation / Profile" button brings you to the
"Predict" window. There are three categories of commands:
- (1) Basic commands: "Load/Save", "Setup" and "Close" buttons
- (2) Evaluate commands: "Chou-Fasman", "Protein profile" and
"Amphipathic" profile buttons
- (3) View commands:
- (3.1) View graph containing "Chou-Fasman" and "Profile" buttons
- (3.2) View consisting of "Chou-Fasman", "Turn", "Profile", and
"Helical wheel" buttons
Chapter 5.8.2.1 Basic (File) commands
"Load/Save" button:
This option allows you to load (open) a file containing a protein
sequence, save the protein sequence in a file, and delete any file (not
only those containing protein sequences).
To load a protein sequence, click on the selected file in the current
path and directory that contains the amino acid sequence of the
peptide/protein, e.g. TEST.SEQ and click on "Load" button (or double
click on selected file). The name of the protein (but not the name of
the file) and its amino acid sequence appears on the screen. The
default directory is the working directory (e.g. C:\PCOM), it can be
changed by double clicking on the new path from the "Load/Save"
window.
| Figure 18 |
|
| Screen Capture of Setup Pop-up
Window |
To save the protein sequence in a file (e.g. after editing the sequence
or writing a new sequence), type the name of the file and click on the
"Save" button. When using a name that already exists on the disk, the
old file will be overwritten by the contents of the file currently residing
in the memory. However, you will be warned about the possibility of
loosing previously stored information.
To delete a file, click on the selected file (or type its name in the
File name window), and click on the "Delete" button. You will be asked to
confirm the operation.
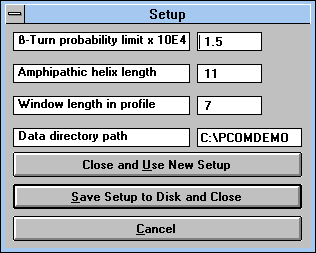
"Setup" (See Figure
18) button:
This command enables one to change the length of the window in the
protein profile, the length of the amphiphilic helix, the probability limit
for beta-turn formation, and the path from which data are read. You can
use the new values temporarily ("Close and Use New Setup"
button), save them to disk ("Save Setup to Disk and Close") or
cancel the setup ("Cancel" button).
"Close" button:
This command returns you to the Peptide Companion main menu.
The currently loaded protein sequence and results of calculation will
be lost.
Editing a sequence:
The sequence residing currently in the memory can be edited. Peptide
Companion uses the standard Windows editor including features
Copy, Delete, Paste, and Undo. For details see your Windows
manual.
Chapter 5.8.2.2 Evaluate commands
| Figure 19 |
|
| Screen Capture of
Chou-Fasman Method |
In the "Predict" window you can evaluate protein profiles, amphiphilic
profiles, and conformational parameters according to the
Chou-Fasman method.
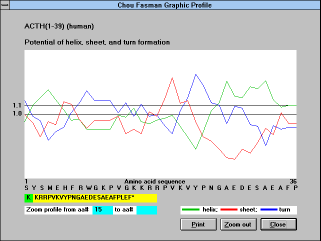
Chou-Fasman parameters
("Chou-Fasman" (See Figure
19) button):
Conformational parameters are evaluated by the Chou-Fasman
algorithm, one of the most common secondary-structure prediction
methods (Chou, P.Y., and Fasman, G.D. (1978) Adv.Enzymol. 47,
46-148). In this method, each amino acid has been assigned a
conformational potential for a-helix, b-sheet, and b-turn formation,
derived from the frequency of its occurrence in the particular
secondary structure. Then, the mean potential of all consecutive
tetrapeptides for all three conformational states (Ph, Ps, and Pt) is
calculated. As a measure of propensity to form a b-turn, an extra
parameter, the positional probability of b-turn occurrence, p, is
calculated by multiplying empirical constants for four consecutive
amino acids. According to Chou and Fasman, the tetrapeptide tends
to form a b-turn when p 0.75 - 1x10-4.
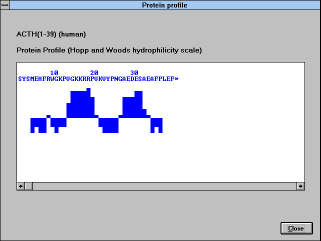
Protein profile ("Protein Profile" button):
Nine different protein profiles can be evaluated. Each amino acid is
assigned a numerical parameter value according to the selected
scale. The program determines the mean value of a seven-peptide
window (default value) moving along the protein sequence. The
profiles are normalized for the purpose of comparing different sets of
parameters. The mean hydrophilicity (or accessibility, etc.) over the
entire protein is calculated and a zero value is arbitrarily set at this
average. The values +1 and -1 are set for the maximum and minimum
local hydrophilicity, respectively.
| Figure 20 |

|
| Screen Capture of Scales Selection
Window |
The following scales are available:
- Parker et al. hydrophilicity scale
J.M.R.Parker, D.Guo, and R.S.Hodges, Biochemistry 25, 5425 (1986).
- Janin accessibility scale
J.Janin, Nature (London) 277, 491 (1979).
- Bulk hydrophobic scale
P.Manavalan and P.K.Ponnuswamy, Nature (London) 275, 673
(1978).
P.A.Karplus and G.E.Schulz, Naturwissenschaften 72, 212 (1985).
- Hopp and Woods hydrophilicity scale
T.P.Hopp and K.R.Woods, Proc.Natl.Acad.Sci. U.S.A. 78, 3824
(1981).
- Fraga global scale
S.Fraga, Can.J.Chem. 60, 2606 (1982).
- Welling et al. antigenicity scale
G.W.Welling, W.J.Weijer, R.van der Zee, and S.Welling-Wester,
FEBS Lett. 188, 215 (1985).
- Hopp acrophilicity scale
T.P.Hopp, in Synthetic Peptides in Biology and Medicine (K.Alitalo,
P.Partanen, and A.Waheri, eds.), p.3. Elsevier, 1985.
- Kyte and Doolittle hydropathy scale
J.Kyte and R.F.Doolittle, J.Mol.Biol. 157, 105 (1982).
- Novotny large sphere accessibility scale
J.Novotny, M.Handschumacher, and R.E.Bruccoleri, Immunol. Today
8, 26 (1987).
You can also define your own scale. Pressing
"Edit Custom Scale" (See
Figure 20)
will let you introduce new parameters, which can be saved for later
use ("Load/Save" button). Above defined scales are stored
independently on the disk (extension *.scl) and can be loaded and
modified. However, we strongly recommend saving newly defined
scales under different names to avoid possible confusion.
Amphipathic profile ("Amphipathic" button):
This subroutine evaluates the amphipathicity of all consecutive protein
fragments of a selected length (default value 11). Using the scale
selected (ten scales available), the hydrophilicity vector is calculated
and normalized in the same way as described for the protein profiles.
The highest value of local amphipathicity indicates the most
hydropathic segment of the protein. The helical amphipathicity has
been shown to correlate with the localization of T-cell determinants
(Margalit H. et al. J.Immunol. 138, 2213 (1987)).
| Figure 21 |
|
| Screen Capture of Protein Profile
Window |
Chapter 5.8.2.3 View commands
To inspect results obtained in the Evaluate procedure, click on the
appropriate button and profiles or parameters will appear on the
screen.
View graph: Clicking on the "Profile" (protein profile or amphipathic
profile) or "Chou-Fasman" button will show you results in a graphical
representation. You can zoom in on any portion of the protein: select
the first amino acid of the zoomed window by mouse (the aa
numbering appears in the window below the profile), click and hold the
left mouse button, move the mouse to the last amino acid of the
zoomed window, and release the mouse button. The "Zoom out"
button will show you the whole protein.
| Figure 22 |
|
| Screen Capture of Helical Wheel
Window |
View: Click on the "Chou-Fasman" button and a table with Chou-Fasman
parameters will appear on the screen. Use the scroll bar to
see any part of the protein. Since B-cell determinants very often reside
in those parts of a protein molecule that have a high tendency to form
a b-turn, the Chou and Fasman algorithm can be used for the
prediction of potential B-cell determinants. Click on the "Turn" button
and the program output lists only tetrapeptide sequences having a
probability of b-turn occurrence greater than a preselected limit,
usually 1.5x10-4 (default value).
The "Profile" button will show you a segment of protein or
amphipathic profile in the bar graph form. This is useful if you want to
see the full profile in the same scale together with the amino acid
sequence. Use the scroll bar to move along the protein sequence.
This option can display only 1000 amino acids.
| Figure 23 |
|
Screen Capture of 3D Helical
Wheel
Window |
An example of an output:
(See Figure 21)
Normalized profile: each bar represents the mean value of a
heptapeptide (default value) window centered at the 4-th amino acid.
The bars extend upwards when the local hydrophilicity is greater than
average, and vice versa.
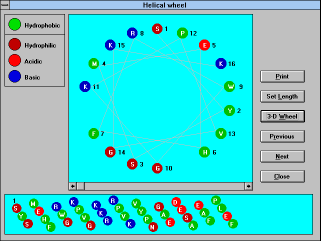
The "Helical wheel" (See
Figure 22) button will show you a standard representation
of the helix in an axial view (default length 18 can be changed
permanently by pressing the "Setup" button and saving the change
after editing the "Setup" window, or temporarily by pressing "Set
Length" button) and in an unwrapped tubular view (standard length
50 amino acid residues). Hydrophobic amino acids are in green
circles, polar ones are in red (acidic), blue (basic), and/or brown
(other) circles. Use the "Next" or "Previous" buttons to walk along
the protein. For fast changes use the horizontal scroll bar. The
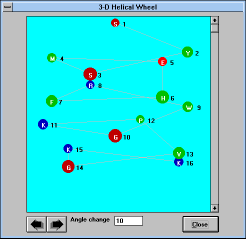
"3D Wheel" (See Figure
23)
shows helical arrangement in three dimensional space. The
view can be turned by defined (editable) increments by clicking on the
arrows below the picture box.
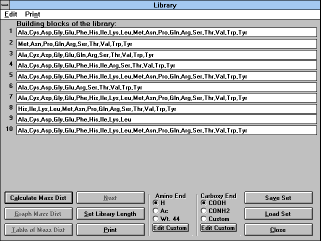
Chapter 5.9. Library
("Library" (See Figure
24) Button)
| Figure 24 |
|
| Screen Capture of Library
Window |
Calculates and graphs the mass distribution in a peptide (or
non-peptide) library.
Distribution of the masses is calculated using integer values of
building block masses and it does not take into account the isotopic
distributions. It can be used for rough evaluation of the quality of
prepared library (Andrews et al., in Techniques in Protein Chemistry,
Vol. V, p. 485, Academic Press, Orlando 1994) or for library design.
The proper selection of library building blocks can simplify mass
spectroscopic evaluation of the results.
New building blocks can be defined for library construction. Care must
be taken not to use an abbreviation already defined for protecting
groups or other structural features. If the newly defined abbreviation
should be made permanent, it has to be saved when you exit the
program.
Clicking on the "Library" button will bring up a dialog box asking you
for the length of the library you wish to randomize. The default is 5
(minimum is 2 and maximum is 30). After you click "OK", the
"Library" window will appear.
| Figure 25 |
|
Screen Capture of Graph of
Mass
Distribution Window |
It uses the following command buttons:
the "Calculate Mass Dist" button (calculates the mass distribution),
the "Graph Mass Dist" button (shows the graph of mass distribution),
the "Table of Mass Dist" button (shows the table of mass
distribution), the "Save Set" button (saves the current set of amino
acids (building blocks) used for the prediction of mass distributions),
the "Load Set" button (loads a set of amino acids (building blocks)
used for the prediction of mass distributions), the "Next / Previous"
buttons (shows next or previous screens of blocks used for
randomization if you set the length of library to more then 15), the
"Print" button (prints the current set of building blocks), the "Set
Library Length" button (sets the length of the library and it also puts
the default set of amino acids in each randomization), and the
"Close" button (returns to the "Main" window).
| Figure 26 |
|
| Screen Capture of Digest Window |
Clicking on the "Table of Mass Dist" button will show the "Table of
Mass Distribution" window. In this window all possible molecular
weights generated in the library, together with the frequency of their
occurence are displayed. In the lower part of the table the highest,
lowest and average values are compiled. You can also save the table
("Save" button) for use in other spreadsheet programs (e.g. Excel,
Quattro Pro) as a text (.TXT) file. This feature can be used for that
purpose only as the table cannot be loaded in Peptide Companion.
Use the "Save Set" button in the "Library" window instead. For
printing the table see 6.3.7.
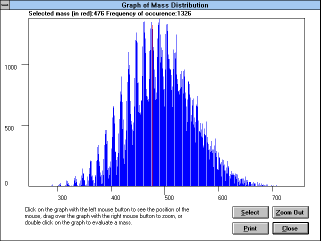
When you press the "Graph Mass Dist"
(See Figure 25)
button, the "Graph of Mass
Distribution" window appears. In this window you can press the right
mouse button to see the coordinates of the cursor, double click on the
graph and select a mass to show its value and frequency of its
occurrence in the library (this can also be done with the "Select"
button), or drag the left mouse button over the graph to zoom (press
"Zoom Out" button to zoom out).
| Figure 27 |
|
| Screen Capture of Switch Boxes
Function |
Chapter 5.10 Enzymatic or chemical degradation of
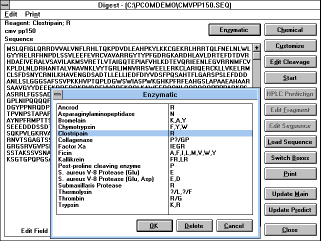
peptides and proteins ("Digest"
(See Figure 26) button)
Proteins or peptides can be submitted to simulated enzymatic or
chemical (or combined) degradation. Loading this window brings
selected peptides to the sequence edit field. When no peptide has
been selected, an empty field will be displayed. A new sequence can
be loaded by pressing the "Load Sequence" button. Pressing
"Enzymatic", "Chemical", "Customize", or "Edit Cleavage" will let
you select the reagent or customize the cleavage specificity. The
number of expected fragments and their list would be shown in the
fragment list.
| Figure 28 |
|
Screen Capture of HPLC Prediction -
Digest Window |
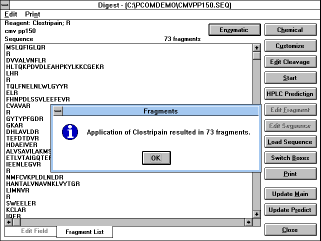
You can switch between the edit field and the fragment list ("Switch
Boxes" button or click on the tab on the bottom of the screen). After
the fragment is selected (by clicking on it), it can be loaded into the
edit field ("Edit Fragment" button) and can be edited for further
cleavage (by different reagent) or it can be loaded into the main
window or predict window ("Update Main" or "Update Predict"
buttons) and to any application from there. The "Customize" button
allows addition of any cleavage reagent and saving it in the database.
The "Digest" window uses the following command buttons.
"Enzymatic" / "Chemical" buttons load reagents from disk. The
"Customize" button enables you to write your own reagent and save
it to the disk as enzymatic, or chemical reagent (requires your
password).
| Figure 29 |
|
| Screen Capture of Find Pop-up
Window |
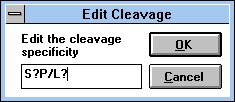
The "Edit Cleavage" button displays a dialog box in which
you type the cleavage specificity which you want to use for the
degradation. This definition cannot be saved. If you want to use a
defined specificity in the future, use the "Customize" button for
saving. The "Start" button starts the cleavage. Choosing any
cleavage specificity using the buttons above will start the cleavage
automatically. The "Edit Fragment" button moves the selected
fragment from the fragment list to the edit field. By starting cleavage,
the originally loaded sequence will be replaced by this fragment and
the old sequence will be cleared.
| Figure 30 |
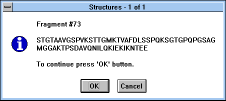
|
Screen Capture of Structures
Pop-up Window |
The "Edit Sequence" button puts
the sequence last used for cleavage or last loaded into the edit field.
The "Load Sequence" button loads the sequence in NIH format. For
example see 5.8.1.
The "Switch Boxes" (See
Figure 27) button switches between the
edit field and the list of fragments. The "Print" button prints fragments
and sequences. The "Update Main" / "Update Predict" buttons
place the selected fragment or the fragment in the edit field (if the edit
field is on top) into the "Main" or "Predict" window. The "Close"
button returns to the "Main" window.
| Figure 31 |
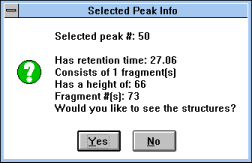
|
Screen Capture of Selected
Peak
Info Pop-up Window |
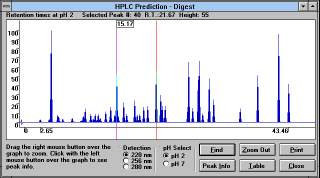
"HPLC Prediction" (See
Figure 28)
button predicts the elution order of fragments on
the reversed phase column. This prediction uses algorithm of Guo et
al. (see section 5.5)
and all limitations discussed earlier apply here as
well. You can choose HPLC with detection at different wavelengths,
using different pH of mobile phase, you can zoom into the crowded
area of the trace, or you can
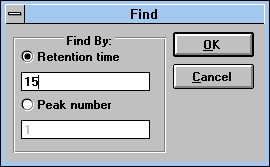
select (See
Figure 29) the
information (See Figure
30)
about any peak (See
Figure 31).
Chapter 5.10.1
Cleavage specificity nomenclature (See Figure
32)
| Figure 32 |
|
Screen Capture of Edit
Cleavage Pop-up Window |
The one-letter code is used in the definition of specificity. The
single letter indicates that the peptide will be cleaved always after this
residue. The specified sequence has the same meaning. Cleavage
between defined residues is specified by the "/" symbol between the
specified residues. The unspecified amino acid is symbolized by "?".
Multiple specificity is defined by putting a comma (",") between the
specificities.
Examples:
- S,P Cleavage after serine or proline
- S/P Cleavage of all -Ser-Pro- sequences
- S?P/L? Cleavage between sequences Ser-Xxx-Pro and Leu-Xxx
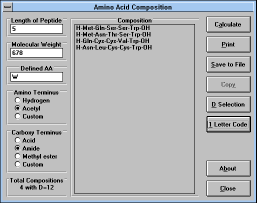
Chapter 5.11 Prediction of amino acid composition
"AA Composition from M.W."
(See Figure 33) button
| Figure 33 |
|
Screen Capture of Amino Acid
Composition Window |
You can calculate probable amino acid composition of peptides
composed of natural amino acids based on their molecular weight. To
make the calculation feasible with present computer technology,
molecular weight is calculated using integer values of monoisotopic
compositions. The calculation is performed for the defined length of
the peptide (Length of Peptide window). If you want to evaluate all
possible peptide lengths, you have to calculate the compositions for
each length separately. Long peptides have usually unreasonably high
number of possible compositions and it is a good idea to narrow the
search down by defining some of the amino acids in the peptide (type
their list in Defined AA window (you can use single letter or three
letter code)). Selection of possible compositions can be further
simplified by measuring number of exchangeable protons in the
peptide molecule (enter the "D Value" -- the number of exchangeable
protons --
("D Selection" (See Figure
34) button)
which can be obtained by measuring
mass spectrum of the sample dissolved in D2O -- see Sepetov et al.
Rapid Comm. Mass Spectrom. 7, 58 (1993)).
| Figure 34 |
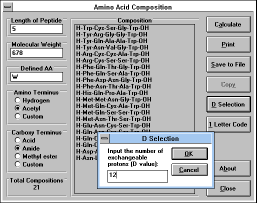
|
Screen Capture of D Selection
Pop-up Window |
It is not possible to distinguish between Ile (I) and Leu (L) , and Lys
(K) and Gln (Q) and therefore only L and Q are used in the calculations.
If you want to calculate composition of cyclic peptide, you must
linearize it, i.e. add the molecular weights of molecules subtracted
during cyclization (e.g. +2 for disulfides, + 18 for lactams).
Fields with blue background signalize that recalculation using newly
defined parameters was not performed. Parameters which can be
changed are: (i) Length of peptide; (ii) Molecular weight; (iii) Defined
amino acids; (iv) Amino terminal group (default H, choices Ac and
Custom); (v) Carboxy terminal group (default OH, choices NH2, OMe,
and Custom). After selection of Custom option you are prompted to
define molecular weight of terminal group and number of
exchangeable protons in this group. (If you will not be using D
Selection, you don't have to bother defining the last value.)
Any calculated composition can be transferred in the form of
sequence (generated by pressing "3 Letter Code" button) into any
other window ("Copy" button). Calculated amino acid compositions
can be saved in the file ("Save to File" button), or printed ("Print"
button). If the number of possible compositions exceed the available
memory space (usually above 4500 compositions), only fraction of
compositions is reported. (However, we believe that information of this
size is not valuable anyway.)
Chapter 5.12 Quit ("Exit" button)
Leaves the program. Program will confirm your intention to quit and it
will ask you whether you want to save any changes to abbreviation
definitions you may have made during the session. You can save the
changes only if you know your password.
Chapter 6 Printing the results
Chapter 6.1 Two ways to print in Peptide Companion
- 1. Regular printing (Default): Every new print job is done
separately (one print job can have several elements). If you are using
Regular printing, the information will print immediately on the whole
page, even if it doesn't take as much space. Graphs (and any other
information that can be printed from the "Predict" window), however,
are printed on a separate sheet of paper every time.
- 2. Consecutive printing: If you are using Consecutive printing,
the computer doesn't print anything until you choose "Print Memory"
in the "Print" menu. In this way you save paper, because you can use
one page to print several outputs. The "Predict" window output is
always printed on a separate sheet of paper, but not printed until you
choose "Print Memory". If you switch off Consecutive printing, the
computer will automatically print everything in the memory.
You can switch between Consecutive and Regular printing on any
window that has the "Print" menu. There are three items in the
"Print" menu, "Consecutive Printing", "Print Memory", and "Paper
Orientation". If "Consecutive Printing" is unchecked "Regular
printing" is on and "Print Memory" will be disabled. When you
check "Consecutive Printing", Consecutive printing will be on and
"Print Memory" will be enabled. When you switch the Consecutive
printing on or off the change will be done in "Print" menus in all
windows.
Chapter 6.2 Changing the paper orientation
You can change the paper orientation by selecting the "Print" menu,
choosing "Paper Orientation" submenu and choosing the orientation.
When "Consecutive Printing" is enabled the program will print the
memory when the orientation is changed.
Chapter 6.3.1 EA, AAA, M.W. window
First select what part you want to print. There are three checkboxes in
the "Print" frame: "M.W./Summary" - this option prints molecular
weight and summary formula; "AAA" prints the amino acid analysis;
"EA" prints the elemental analysis. Then press the "Print" button.
Chapter 6.3.2 Correction of Elemental Analysis window
The printing procedure is very similar to the previous one. There are
two option buttons: "All Fits" prints all fits if you calculated the best fit;
"Selected Fit" prints just the selected fit. There is also a "Results"
checkbox; if it is checked, it will print the results obtained by
calculation. Press the "Print" button.
Chapter 6.3.3 HPLC Prediction window
When you press the "Print" button in the "HPLC Prediction" window
the computer will ask you if you want to "Print All", which means that
the values plus the graph are going to be printed (this option cannot
be printed into memory and will be printed immediately), or "Only
Values" which means that only the peptide formulas plus the values
are going to be printed.
Chapter 6.3.4 MS Fragmentation window
You can print "All Fragments" or just the "Selected Fragment" by
choosing an option in the "Print" frame. You can choose to print All
Masses by checking the "All Masses" check box; they will be printed
as the last information.
Chapter 6.3.5 Prediction of Difficult Sequence window
Refer to printing Graphs in the "Predict" window.
Chapter 6.3.6 Predict window
All graphs in the "Predict" window that have a "Print" button can be
printed; you can also print the Chou-Fasman parameters. If you zoom
the graph, it will be printed zoomed.
Chapter 6.3.7 Library window
In the "Library" window you can print the set of building blocks by
pressing the "Print" button. In the "Table of Mass Distribution"
window you can print the table by pressing the "Print" button. In the
"Graph of Mass Distribution" window you can print the graph (as
seen on screen) also by clicking the "Print" button (this is the same
as in the "Predict" window).
Chapter 6.3.8 Digest window
You can print the fragments of the peptide by pressing the "Print"
button. The computer will ask you whether you also want to print the
whole sequence.
Chapter 7 Troubleshooting
| 1 |
Problem: |
| This manual is not clear enough about a certain
point or does not contain the information you want. |
| Solution: |
| See Help for more detailed information. |
| 2 |
Problem: |
| Setup program stops, saying it cannot copy
VBRUN300.DLL. |
| Solution: |
| If you already have a copy of VBRUN300.DLL in your
"\windows\system" directory try renaming it, to VBRUN300.000 for
example, (do not delete it) and run the setup program again. To do
this start File Manager and go to your windows system directory,
usually "C:\WINDOWS\SYSTEM", then locate the file
"VBRUN300.DLL" and select it. From the "File" menu choose the
"Rename..." item. Type in the new name and press the "OK" button. |
| 3 |
Problem: |
| Message "Critical Datafile Missing." |
| Solution: |
| Run setup program again without upgrading any
datafiles. |
| 4 |
Problem: |
| When you scroll on a scrollbar to see data not
currently displayed and they appear distorted, or when a graphical
representation, like helical wheel, is cut into many small fragments. |
| Solution: |
| Try using standard VGA or SuperVGA display driver.
Some drivers can cause problems. |
| 5 |
Problem: |
| Some text results do not fit the boxes or are
illegible. |
| Solution: |
| Maybe your display driver does not use standard
Windows fonts, use another font size or a different driver. |
| 6 |
Problem: |
| "Out of memory" message when starting. |
| Solution: |
- I. Quit all unnecessary applications.
- II. Start "Peptide Companion Startup" and turn off the "Load all
windows" checkbox, then check the "Unload windows not in use"
checkbox and press the "OK" button. Restart Peptide Companion.
|
| 7 |
Problem: |
| "Start" button in Digest window is not enabled. |
| Solution: |
| No sequence or reagent was specified. When you
specify a reagent start button is pressed automatically. |
| 8 |
Problem: |
| I have further problems, questions or
suggestions. |
| Solution: |
| If you have any further problems, questions or
suggestions contact:
CSPS Pharmaceuticals
6161 Arnoldson Ct.
San Diego, CA 92122-2115
U.S.A.
FAX: (858) 550-9666
Email: csps@5z.com |
|
Disclaimer:
CSPS PHARMACEUTICALS SHALL HAVE NO LIABILITY WITH RESPECT TO ANY LOSS
OR DAMAGE DIRECTLY OR INDIRECTLY ARISING OUT OF THE
USE OF THE DISK, THE PROGRAMS OR THE DOCUMENTATION.
WITHOUT LIMITING THE FOREGOING, CSPS SHALL NOT BE
LIABLE FOR ANY LOSS OF PROFIT, INTERRUPTION OF
BUSINESS, DAMAGE TO EQUIPMENT OR DATA, INTERRUPTION
OF OPERATIONS OR ANY OTHER DAMAGE, INCLUDING BUT
NOT LIMITED TO DIRECT, SPECIAL, INCIDENTAL OR
CONSEQUENTIAL DAMAGES.
Appendix A Command Buttons
- Main window
- List Peptides Button - By clicking on it you can choose a Peptide Dbase.
- Write From Scratch Button / Edit Peptide Button - Shows the Peptide Edit Tools.
- Three Letter Code Button - Translates one letter code to three letter code.
- EA, AAA, M.W. Button - Calculates elemental analysis, amino acid analysis and the molecular weight of a peptide.
- HPLC Prediction Button - Calculates RP HPLC retention times.
- MS Fragmentation Button - Fragmentation for mass spectrometry.
- Difficult Sequences Button - Prediction of synthetically difficult sequences.
- Conformation / Profile Button - Displays the Predict window.
- Library Button - Displays the Library window.
- Digest Button - Displays the Digest window.
- Delete Peptide Button - Deletes Peptide from the database.
- List Specifications - Lists Peptides according to custom specifications.
- New Abbreviation Button - Adds custom abbreviation to the list.
- Edit Abbreviation Button - Edits selected abbreviation.
- Exit Button - Exits Peptide Calculator.
- Predict window
- Load / Save Button - Loads and saves proteins.
- Setup Button - Sets defaults for Predict window.
- Close Button - Returns to Main window. All calculations are lost.
- Evaluate Commands - Lets you choose the following:
- Chou-Fasman Button - Performs the predictions according to Chou and Fasman.
- Protein Profile Button - Lets you choose the appropriate scale, or define a new one.
- Amphipathic Button - Lets you choose the appropriate scale, or define a new one.
- View Commands
- View Graph
- Chou-Fasman Button - graph representation of the calculated data is shown.
- Profile Button - shows calculated data in linear graph form.
- View
- Chou-Fasman Button - tabular representation of the calculated data is shown.
- Turn Button - shows table of probable turns in the protein.
- Profile Button - shows calculated data in bar graph form.
- Helical Wheel Button - shows helical wheel representation of the protein or peptide.
- EA, AAA, M.W. window
- Recalculate Button - Recalculates all calculations with the edited peptide.
- Add AcOH ... / Fit to Found Buttons - Addition of various molecules
(Add AcOH ... button) and calculation of probable composition of the lyophilizate (Fit to Found button).
- Other Elements Button - Displays composition of all elements in the sample.
- Save Formula Button - Saves Formula of the peptide.
- Print Button - Prints results.
- Close Button - Returns to Main window.
- Correction of Elemental Analysis window
- Best Fit Button - Calculates the best fit and all satisfactory results.
- Print Button - Prints results.
- Calculate Button - Calculates elemental analysis with added molecules.
- Close Button - Returns to EA, AAA, M.W. window.
- HPLC Prediction window
- Predict Button - Calculates HPLC Prediction.
- Print Button - Prints results.
- Set Standard Time Button - Allows you to input the experimentally observed retention time of the standard peptide.
- Clear Trace Button - Same as Predict Button, but clears the graph first.
- Load Standard Button - Loads the formula of a standard peptide.
- Load Sample Button - Loads the formula of a sample peptide.
- Close Button - Returns to Main window.
- MS Fragmentation window
- Calculate Button - Recalculates MS fragmentation after editing the sample.
- Found Button - Compares results from spectral measurement with theoretical data.
- Print Button - Prints results.
- Close Button - Returns to Main window.
- Library window
- Calculate Mass Dist Button - Calculates the mass distribution.
- Graph Mass Dist Button - Shows the graph of mass distribution.
- Table of Mass Dist Button - Shows the table of mass distribution.
- Save Set Button - Saves the current set of amino acids (building blocks) used for the prediction of mass distributions.
- Load Set Button - Loads a set of amino acids (building blocks) used for the prediction of mass distributions.
- Next / Previous Buttons - Shows next or previous screens of blocks used for randomization if you set the length of library to more then 15.
- Print Button - Prints the current set of building blocks.
- Set Library Length Button - Sets the length of the library. It puts the default set of amino acids in each randomization.
- Close Button - Returns to Main window.
- Digest window
- Enzymatic / Chemical Buttons - Loads reagent from disk.
- Customize Button - Enables you to write your own reagent and save it to the disk as enzymatic, or chemical (requires your password).
- Edit Cleavage Button - Displays a dialog box in which you type custom cleavage specificity. Cannot be saved, use Customize button for saving.
- Start Button - Starts the cleavage. Choosing any cleavage specificity using the buttons above will start the cleavage automatically.
- HPLC Prediction Button - Calculates retention times of fragments on reversed phase column.
- Edit Fragment Button - Puts the selected fragment from the fragment list to the edit field. By starting cleavage the sequence will be replaced by this fragment and the old sequence will be cleared.
- Edit Sequence Button - Puts the sequence last used for cleavage or last loaded into the edit field.
- Load Sequence Button - Loads Sequence in NIH format.
- Switch Boxes Button - Switches between the edit field and the list of fragments.
- Print Button - Prints fragments and sequence.
- Update Main / Update Predict Buttons - Put the selected fragment or the fragment in the edit field (if the edit field is on top) in the Main or Predict window.
- Close Button - Returns to Main window.
- Amino Acid Composition window
- Calculate Button - Performs the calculation.
- Print Button - Prints the list of possible amino acid compositions.
- Save to File Button - Saves the results in either one- or three-letter code (in the form of sequence).
- Copy Button - Copies the selected composition from the list to clipboard.
- D Selection - Selects the compositions from the list which fulfil the condition of number of exchangeable protons.
- 3 Letter Code Button - Translates one letter code list of amino acids to peptide sequence written in the three letter code.
- 1 Letter Code Button - Performs just the opposite.
- About Button - Describes function of this window.
- Close Button - Returns to Main window.
In all windows Close or Cancel button will return to the previous
window. Cancel button cancels all operations on the windows closed,
Close button does not.
Appendix B Menus